 2016, Vol. 43
2016, Vol. 43文章信息
- 袁忠民,曾敏灵,唐晓梅,伍燕娜,何国振.
- YUAN Zhongmin, ZENG Minling, TANG Xiaomei, WU Yanna, HE Guozhen.
- 组蛋白去乙酰化酶抑制剂对胶质瘤细胞增殖及MKK7表达的影响
- Effects for Histone Deacetylase Inhibitors on Proliferation and MKK7 Expression in Glioma Cells
- 肿瘤防治研究, 2016, 43(01): 1-5
- Cancer Research on Prevention and Treatment, 2016, 43(01): 1-5
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2016.01.001
-
文章历史
- 收稿日期: 2015-04-09
- 修回日期: 2015-09-23
引用本文 |

神经胶质瘤是中枢神经系统最常见的原发性肿瘤,恶性增殖和高侵袭性是其主要特点,亦是造成高复发率的首要原因[1, 2]。基于分子生物学和遗传学的研究表明,JNK/c-Jun通路的信号异常激活对胶质瘤细胞的恶性增殖、侵袭起重要作用[3, 4, 5, 6],但上游信号分子尚不清楚,而且干预这个通路的活性进而抑制胶质瘤生长的药物知之甚少。
本研究采用小分子干扰、BrdU掺入实验、Western blot、转染等技术,检测上游激酶MKK7是否为调控JNK/c-Jun活性及胶质瘤细胞增殖的关键分子;用近年来备受关注的抗肿瘤化合物组蛋白去乙酰化酶抑制剂[7, 8, 9]包括TSA、SAHA、VPA、M344处理胶质瘤细胞U251,检测这些药物能否影响MKK7的表达及功能。
1 材料与方法 1.1 材料U251胶质瘤细胞株购自上海细胞生物学研究所中国科学院细胞库。胎牛血清购于美国Hyclone公司。培养液高糖DMEM、转染试剂RNAiMAX均购于美国Invitrogen公司。MKK7 siRNA、空白siRNA购自上海Gene Pharma公司。细胞实验所用24和6孔板购自美国Corning公司。ECL发光试剂、Hoechst 33258、M344、SAHA、TSA、VPA、BrdU、anti-BrdU抗体均购于美国Sigma公司。anti-MKK7抗体(#1949-1)购自美国Epitomics公司,anti-p-JNK(T183/Y185)(#9251S)、anti-p-c-Jun(S73)(#9164)购于美国CST公司,anti-c-Jun(#sc-1694)、anti-JNK(#sc-7345)均购于美国Santa Cruz公司。HRP标记抗兔、Cy3标记抗鼠购于美国JacksonRes公司。PVDF膜购于美国Millipore公司。MKK7-siRNA1小分子片段序列正义链:5’-CCAACACGGACGUCUUCAU-3’,反义链:5’-AUGAAGA CG UCCGUGUUGG-3’; MKK7-siRNA2小分子片段序列正义链:5’-GCUGGCAACAGGACAGUUU-3’,反义链:5’-AAACUGUCCUGUUGCCAGG-3’。
1.2 方法 1.2.1 细胞培养参考已有研究[10]。将U251置于10%胎牛血清、高糖DMEM液体培养液,37℃、5%CO2及饱和湿度条件下培养,每3~5天用胰酶消化传代1次.取对数生长期细胞进行实验。
1.2.2 脂质体介导MKK7-siRNA转染参考已有研究[10]。于6孔板内接种对数生长期U251细胞,细胞密度为每孔2×105个,培养液为不含抗生素的DMEM培养液。待细胞生长至60%~80%融合度时,更换为新鲜培养液10%胎牛血清的DMEM培养液,按转染试剂RNAiMAX转染方法转染MKK7-siRNA1、MKK7-siRNA2和空白对照siRNA,放入37℃培养箱培养48 h后做Western blot检测,检测U251转染细胞中MKK7的表达。
1.2.3 Westernblot检测 取对数生长期U251细胞,以每孔5×105个接种于6孔板中,待细胞生长到70%融合度时,加入HDACIs(1 μmol/L M344、10 μmol/L SAHA、0.5 μmol/L TSA、6 mmol/L VPA),设置对照孔(0.1% DMSO)。8 h后每孔PBS 2 ml漂洗2遍,每孔加入1 ml IP细胞裂解液(50 mmol/L Tris,HCl pH8.0,150 mmol/L NaCl,1% Triton×100,100 μg/ml PMSF),10 min后收集各组细胞。按照IP裂解液法提取细胞总蛋白,并进行BCA法测定蛋白浓度。灌制4%集成胶和8%分离胶。200 V电压电泳45 min。100 V电压转膜60 min。5%脱脂奶粉室温封闭1 h。一抗(anti-MKK7 1:1 000,anti-GAPDH 1:5 000,anti-p-JNK 1:1 000,anti-p-c-Jun 1:1000, anti-c-Jun 1:1 000,anti-JNK1:1 000)孵育过夜(4℃),HRP标记抗兔室温孵育1 h。ECL化学发光后曝光。
1.2.4 BrdU掺入实验(BrdUIncorporation) 参考已有研究[10]。取处于对数生长期的U251细胞以每孔2×104个接种于24孔板,待细胞生长至50%融合度时,更换为新鲜的、含10%胎牛血清的DMEM培养液,按转染试剂RNAiMAX转染方法转染MKK7-siRNA1、MKK7-siRNA2和空白对照siRNA,每组设3个复孔,37℃培养箱培养48 h;终止细胞培养前,加入BrdU(终浓度10 μmol/L),继续培养4 h,弃培养液;PBS洗3次,每次5 min;4%多聚甲醛冰上固定30 min;PBS洗3次,每次5 min;2M HCl室温孵育30 min;PBS洗3次,每次5 min;0.1% TBS/Triton配成3%BSA封闭1 h;anti-BrdU抗体(1:1 000)4℃孵育过夜;PBS洗3次,每次5 min;Cy3(1:200)标记抗鼠二抗孵育1 h;PBS洗3次,每次5 min;Hoechst 33258(5 μg/ml)染核后置于荧光倒置显微镜拍照并计算细胞增殖率。细胞增殖率=BrdU阳性细胞/总细胞数×100%。
取对数生长期U251细胞以每孔5×104个接种于24孔板,每孔500 μl,过夜待细胞贴壁后换液,设0.5 μmol/L TSA处理细胞为实验组,药物溶剂处理细胞为对照组,每组设2个复孔,作用8、12、24 h后用PBS每孔1 ml漂洗2次,每孔加入Hoechst 33258(5 μg/ml)每孔500 μl培养箱孵育15 min; PBS漂洗3次,每次5 min;荧光倒置显微镜拍照(×200)计算细胞增殖率。细胞增 殖率=(实验组细胞数/对照组细胞数)×100%,实验重复3次。其余组蛋白去乙酰化酶抑制剂(1 μmol/L M344、10 μmol/L SAHA、6 mmol/L VPA)处理细胞,计算12 h细胞增殖率,方法同前。
1.3 统计学方法采用SPSS19.0统计软件包进行统计分析。分析值比较采用单因素方差分析,实验结果以均数±标准差表示,Student’s t检验,P<0.05为差异有统计学意义。
2 结果 2.1 转染siRNA对MKK7表达及JNK/c-Jun活性的影响同对照siRNA比较,MKK7-siRNA1和MKK7-siRNA2均显著抑制MKK7表达,并且下调p-JNK和p-c-Jun磷酸化水平,下调c-Jun表达,见图 1。

|
| 1: siRNA control group; 2: MKK7-siRNA1 group; 3: MKK7-siRNA2 group 图 1 Western blot检测小分子干扰对MKK7的表达及JNK、c-Jun活性的影响 Figure 1 Effects of siRNAs on MKK7 expression and JNK, c-Jun activities analyzed by Western blot |
转染对照siRNA的BrdU掺入率为(32±4)%,转染MKK7-siRNA1、MKK7-siRNA2 BrdU掺入率分别为(24±2)%、(23±3)%,与对照组比较差异均有统计学意义(n=3,P均=0.008),见图 2,表明沉默MKK7表达抑制细胞增殖。

|
| 1: siRNA control group; 2: MKK7-siRNA1 group; 3: MKK7-siRNA2 group; *: P=0.008, compared with siRNA control group 图 2 BrdU掺入法检测沉默MKK7表达对U251胶质瘤细胞增殖率的影响 Figure 2 Effect of MKK7 knockdown on proliferation rate of U251 glioma cells detected by BrdU incorporation analysis |
同对照组比较,0.5 μmol/L TSA处理细胞8 h可显著抑制MKK7表达及JNK/c-Jun磷酸化,见图 3。

|
| 1: DMSO; 2: 0.5μmol/L TSA 图 3 Western blot检测TSA处理对MKK7表达及JNK/c-Jun活性的影响 Figure 3 Effect of TSA on MKK7 expression and JNK/c-Jun activities detected by Western blot |
同对照组比较,其他组蛋白去乙酰化酶抑制剂包括SAHA、M344、VPA处理8 h均可显著抑制MKK7的表达,见图 4。

|
| 1: DMSO; 2: 10μmol/L SAHA; 3: 1μmol/L M344; 4: 6mmol/L VPA 图 4 Western blot检测SAHA、M344、VPA对MKK7表达的影响 Figure 4 Effect of SAHA, M344 and VPA on MKK7 expression detected by Western blot |
计数细胞结果显示,与对照组比较,TSA处理8 h增殖率无明显变化(P=0.823),处理12 h显著抑制细胞增殖(P=0.006),处理24 h抑制效应更明显(P=0.002),见表 1。其他几种组蛋白去乙酰化酶抑制剂包括SAHA、VPA、M344处理12 h均能抑制细胞增殖(n=5,P=0.001、0.008、0.002),见表 2。
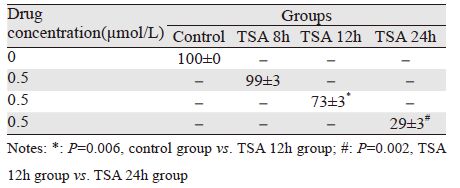 |
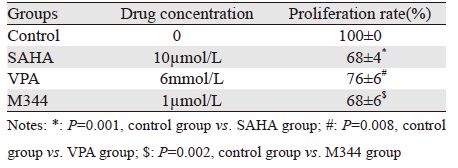 |
本研究中,siRNAs沉默MKK7表达抑制JNK/c-Jun活性以及胶质瘤细胞U251增殖;TSA抑制MKK7表达及JNK/c-Jun活性发生在抑制细胞增殖之前;所有使用的组蛋白去乙酰化酶抑制剂都能抑制MKK7表达和神经胶质瘤细胞增殖。
JNK是有丝分裂原激活蛋白激酶(mitosis-activated protein kinase,MAPK)家族的重要成员,被上游激酶如MKK4或MKK7在183位苏氨酸和185位酪氨酸磷酸化后活性增强,进而激活下游原癌基因如c-Jun表达[11, 12, 13],促进包括胶质瘤[6]、肝癌[11]、前列腺癌[14]等多种肿瘤发生。但在胶质瘤中,是MKK4或MKK7介导JNK/c-Jun磷酸化和激活尚不清楚。利用siRNAs特异沉默MKK7表达,我们证明MKK7介导了JNK/c-Jun激活,而且其活性促进细胞增殖,但MKK4是否也参与了介导还有待进一步研究。
近年来,组蛋白去乙酰化酶抑制剂之所以被关注是缘于其突出的促进肿瘤细胞凋亡或抑制细胞周期效应,而这些效应在正常细胞中的敏感度明显下降[15]。JNK/c-Jun作为一个广受关注的调控肿瘤细胞存活和死亡的应激通路,在多种肿瘤细胞中都发现组蛋白去乙酰化酶抑制剂通过激活JNK/c-Jun诱导细胞凋亡[16, 17, 18]。但本研究表明在胶质瘤细胞U251中,组蛋白去乙酰化酶抑制剂通过抑制MKK7表达及其介导的JNK/c-Jun活性抑制肿瘤生长,即使使用能强烈诱导U251细胞凋亡的、较高浓度组蛋白去乙酰化酶抑制剂,JNK活性依然被抑制,本研究未检测到MKK7/JNK激活,而且三种不同结构的组蛋白去乙酰化酶抑制剂的结果一致,这表明组蛋白去乙酰化酶抑制剂抑制肿瘤的机制具有组织特异性。下一步工作将明确在胶质瘤细胞中组蛋白去乙酰化酶抑制剂抑制MKK7表达的机制。
总之,我们在U251胶质瘤细胞株中首次证明组蛋白去乙酰化酶抑制剂的抗癌机制中包括抑制MKK7表达及下游JNK/c-Jun活性,本研究结果对理解和认知组蛋白去乙酰化酶抑制剂的药理作用机制具有一定理论意义,在我们使用的三种组蛋白去乙酰化酶抑制剂中,SAHA和VPA是FDA批准的临床用药,提示组蛋白去乙酰化酶抑制剂很有希望成为新的抗胶质瘤药物。
| [1] | Alexandru-Abrams D, Jadus MR, Hsu FP, et al. Therapeutic targeting of malignant glioma[J]. Anticancer Agents Med Chem, 2014, 14(8): 1075-84. |
| [2] | Zhou ZL, Bu XY, Yan ZY, et al. Effects of rendezvous chemotherapy compined with three-dimensional comformal radiotherapy in treatment for post-operative malignant glioma[J]. Zhong Liu Fang Zhi Yan Jiu, 2014, 41(4): 374-8. [周志龙, 步星耀, 闫兆月, 等. 脑恶性胶质瘤术后会师化疗同步适形放疗的临床观察[J]. 肿瘤防治研究, 2014, 41(4): 374-8.] |
| [3] | Kitanaka C, Sato A, Okada M. JNK Signaling in the Control of the Tumor-Initiating Capacity Associated with Cancer Stem Cells[J]. Genes Cancer, 2013, 4(9-10): 388-96. |
| [4] | Li JY, Wang H, May S, et al. Constitutive activation of c-Jun N-terminal kinase correlates with histologic grade and EGFR expression in diffuse gliomas[J]. J Neurooncol, 2008, 88(1): 11-7. |
| [5] | Dong L, Ge RM, Qi N, et al. JNK1 knockdown via adenovirus-mediated shRNA inhibited cell proliferation in U87MG gliobalstoma cells[J]. Zhong Liu Fang Zhi Yan Jiu, 2011, 38(7): 767-9. [董林, 葛瑞民, 祁楠, 等. shRNA腺病毒介导的JNK1 RNAi抑制U87MG人胶质瘤细胞的增殖[J]. 肿瘤防治研究, 2011, 38(7): 767-9.] |
| [6] | Cui J, Han SY, Wang C, et al. c-Jun NH(2)-terminal kinase 2alpha2 promotes the tumorigenicity of human glioblastoma cells[J]. Cancer Res, 2006, 66(20): 10024-31. |
| [7] | Bose P, Dai Y, Grant S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights[J]. Pharmacol Ther, 2014, 143(3): 323-36. |
| [8] | Noureen N, Rashid H, Kalsoom S. Identification of type-specific anticancer histone deacetylase inhibitors: road to success[J]. Cancer Chemother Pharmacol, 2010, 66(4): 625-33. |
| [9] | Slingerland M, Guchelaar HJ, Gelderblom H. Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors[J]. Anticancer Drugs, 2014, 25(2): 140-9. |
| [10] | Yuan Z, Gong S, Luo J, et al. Opposing roles for ATF2 and c-Fos in c-Jun-mediated neuronal apoptosis[J]. Mol Cell Biol, 2009, 29(9): 2431-42. |
| [11] | Guo Y, Wang W, Wang J, et al. Receptor for activated C kinase 1 promotes hepatocellular carcinoma growth by enhancing mitogen-activated protein kinase kinase 7 activity[J]. Hepatology, 2013, 57(1): 140-51. |
| [12] | Kaikai S, Yuchen S, Lili J, et al. Critical role of c-Jun N-terminal kinase in regulating bFGF-induced angiogenesis in vitro[J]. J Biochem, 2011, 150(2): 189-97. |
| [13] | Rennefahrt U, Illert B, Greiner A, et al. Tumor induction by activated JNK occurs through deregulation of cellular growth[J]. Cancer Lett, 2004, 215(1): 113-24. |
| [14] | Miyazaki T, Bub JD, Iwamoto Y. c-Jun NH(2)-terminal kinase mediates leptin-stimulated androgen-independent prostate cancer cell proliferation via signal transducer and activator of transcription 3 and Akt[J]. Biochim Biophys Acta, 2008, 1782(10): 593-604. |
| [15] | Nalabothula N, Carrier F. Cancer cells' epigenetic composition and predisposition to histone deacetylase inhibitor sensitization[J]. Epigenomics, 2011, 3(2): 145-55. |
| [16] | Sarkar D, Leung EY, Baguley BC, et al. Epigenetic regulation in human melanoma: past and future[J]. Epigenetics, 2015, 10(2): 103-21. |
| [17] | Rosato R, Hock S, Dent P, et al. LBH-589 (panobinostat) potentiates fludarabine anti-leukemic activity through a JNK- and XIAP-dependent mechanism[J]. Leuk Res, 2012, 36(4): 491-8. |
| [18] | Sharma V, Koul N, Joseph C, et al. HDAC inhibitor, scriptaid, induces glioma cell apoptosis through JNK activation and inhibits telomerase activity[J]. J Cell Mol Med, 2010, 14(8): 2151-61. |