 2014, Vol.41
2014, Vol.41

2.河北医科大学第二医院血液科 血液病河北省重点实验室
2. Department of Hematology, The Second Hospital, Hebei Medical University
急性白血病(acute leukemia,AL)是常见的恶 性肿瘤,JAK/STAT(Janus kinase-signal transducer and activator of transcription signaling pathway)途径 的激活与白血病细胞增殖分化有关,SHP-1是JAK/ STAT信号转导通路的负调控机制之一。SHP-1基因的表达缺失可归因于SHP-1的突变、启动区的甲 基化或SHP-1蛋白合成转录后调节。研究证实,淋 巴瘤和白血病细胞系中存在SHP-1基因表达缺失及 其启动子甲基化[1],5-aza-CdR能诱导这些细胞系 SHP-1基因表达,并抑制肿瘤细胞增殖[2]。As2O3 具 有杀伤白血病细胞、促进白血病细胞凋亡和诱导 细胞分化三重作用,并发挥去甲基化的作用[3]。本 实验以白血病细胞株U937为研究对象,通过实时 定量PCR法,从mRNA水平研究了应用去甲基化 药物5-aza-CdR和As2O3 后,SHP-1、JAK3、TYK2 基因的表达情况,分析了SHP-1基因的表达缺失 的原因及对JAK/STAT通路的影响,为进一步探讨 SHP-1基因和JAK家族基因在白血病发病中的作用 奠定基础。 1 材料和方法 1.1 材料
白血病细胞株U937,为河北医科大学第二医 院血液病研究所试验室保留株。RPMI 1640培养 液和新生胎牛血均购自Hyclone公司。5-aza-CdR 购自美国Sigma公司,用10% RPMI 1640培养液 配成4 mol/L储备液,-20℃保存,稀释终浓度为 200 μmol/L。As2O3 (三氧化二砷,ATO)购自哈 尔滨伊达药业有限公司,应用浓度为500 μmol/L。 TRIzol Reagent购自美国Invitrogen公司。SYBR GreenⅠ购自上海基康生物技术公司。SHP-1上游 引物:5′-GCC TGG ACT GTG ACA TTG AC-3′, 下游引物:5′-ATG TTC CCG TAC TCC GAC TC-3′。JAK3上游引物:5′-CCA CGG TCT GGG AAG TGT TTA G-3′,下游引物:5′-ACG AAT GAC GGC TCG GAA G-3′。TYK2上游引物: 5′-TCA CTG AGT TGC TGG AAC GAG G-3′,下游 引物:5′-CGA AGG TTG GGC GAA AGG AC-3′。 β-actin上游引物:5′-TCA TCA CCA TTG GCA ATG AG-3′,下游引物:5′-CAC TGT GTT GGC GTA CAG GT-3′。引物由上海生工基因技术有限公司合 成,SHP-1的扩增片断长度187 bp,JAK3的扩增 片断长度188 bp,TYK2的扩增片断长度117 bp, β-actin的扩增片断长度155 bp。 1.2 细胞培养
U937细胞悬浮培养于含10%热灭活(56℃, 30 min)的新生胎牛血清、青霉素100 u/ml、链霉 素100 μg/ml的RPMI 1640培养液中。培养条件为 37℃、5%CO2饱和湿度,隔天换液传代一次。取 对数生长期的细胞用于实验。 1.3 药物处理
将对数生长期的细胞转入细胞培养板,每孔1 ×106cells/ml。5-aza-CdR浓度为0.5、1、2 μmol/L, As2O3 的浓度为1、2.5、5 μmol/L,As2O3 1 μmol/L+ 5-aza-CdR 0.5 μmol/L(低浓度组)、As2O3 2.5 μmol/L+5-aza-CdR 1 μmol/L(中浓度组) 、As2O3 5 μmol/L+5-aza-CdR 2 μmol/L(高浓度组) [2]及空白对照组。细胞和药物在37℃、5%CO2饱和湿度 条件下孵育24、48、72 h后收获并洗涤。 1.4 荧光实时定量PCR(RQ-PCR)检测SHP-1、 JAK3、TYK2的基因表达
Trizol提取细胞总RNA,在紫外透射仪上显示 有28S、18S(部分可见到5S)两条rRNA条带,说 明提取的RNA完整。紫外分光光度计定量。反转 录反应体系20 μl,包括细胞总RNA 2 μg,50 u/μl Rnasin 1 μl,500 μg/ml随机引物1 μl,10 mM dNTP 2 μl,5×反转录反应缓冲液4 μl,M-MLV反转录酶 200 u,余用DEPC水补足至20 μl。反应条件为37℃ 中反应60 min,95℃反应5 min后终止反应。cDNA 在-80℃保存或进行PCR扩增。
RQ-PCR反应总反应体系25 μl,包括cDNA 1.5 μl,10 pmol/μl 上、下游引物各1 μl,dNTP 5 mM 0.8 μl,10×buffer 2.5 μl,MgCl2 25 mM 1.5 μl,Taq酶3 u/μl 0.5 μl,20×SYBR Green I 1 μl, 余用DEPC水补足25 μl。SHP-1的扩增条件:94℃ 变性45 s,60℃退火45 s,72℃延伸60 s,35个循 环。最后72℃延伸10 min。JAK3的扩增条件: 94℃变性45 s,60℃退火45s,72℃延伸60s,35个 循环。最后72℃延伸10min。TYK2的扩增条件: 94℃变性45s,60℃退火45 s,72℃延伸60 s,35个 循环。最后72℃延伸10 min。β-actin的扩增条件: 94℃变性45s,58℃退火45s,72℃延伸60 s,35个 循环。最后72℃延伸10 min。 1.5 PCR产物的相对定量分析
通 过 标 准 曲 线 对 检 测 样 品 的 目 的 基 因 (SHP-1、JAK3、TYK2)及管家基因(β-actin) 进行定量,然后用下列公式计算,求得相对值即 为相对表达量。 相对值=2-ΔΔCt,ΔCt=Ct1-Ct2,Ct1:处理样品待 测基因的临界循环数,Ct2:处理样品管家基因的 临界循环数。 1.6 统计学方法
组间比较采用多样本均数比较的方差分析。 采用配对t检验分析单用药与合用药对基因表达的 影响。采用直线回归分析,探讨SHP-1对JAK3、TYK2的影响。所有数据用SPSS.V13.0统计软件分 析处理,P<0.05表示差异有统计学意义。 2 结果 2.1 SHP-1基因在U937细胞中的表达
单独应用As2O3 和5-aza-CdR以及两药合用时, 随着药物浓度的增加和作用时间的延长,SHP-1的 表达水平升高,与未用药的对照组以及各组间两 两比较差异均有统计学意义(P<0.05)。合用药 均比单独用药时的表达水平升高,差异有统计学 意义(P<0.05),见图 1,表 1、2。
 |
1:As2O3 low;2:As2O3 mid;3:3:As2O3 high;4:5-aza-CdR low;5:5-azaCdR mid;6:5-aza-CdR high;7:As2O3 + 5-aza-CdR low;8:As2O3 + 5-azaCdR mid;9:As2O3 + 5-aza-CdR high
图1 SHP-1基因在U937细胞中的表达
Figure 1 SHP-1 mRNA expression in U937 cells |
|
|
表 1 单用As2O3 及联合用药处理后SHP-1基因在U937细胞中表达比较 Table 1 Comparison of SHP-1 gene expression in U937 cells treated with As2O3 and the combination |

|
|
表 2 单用5-aza-CdR及联合用药处理后SHP-1基因在U937细胞中的表达比较 Table 2 Comparison of SHP-1 gene expression in U937 cells treated with 5-aza-CdR and the combination |

随着药物浓度的增加和作用时间的延长, J A K 3的表达水平下降,与未用药的对照组比 较差异均有统计学意义(P<0.05)。除中浓度 (2.5 μmol/L)时,48与72 h差异无统计学意义 (P=0.111)外,其余各浓度在各时段的表达变 化经两两比较差异均有统计学意义(P<0.05)。 在三个时段(24、48、72 h)内低、中、高浓度 间的表达变化经两两比较,差异均有统计学意义 (P<0.05),见图 2、表 3。
 |
1:As2O3 low;2:As2O3 mid;3:3:As2O3 high;4:5-aza-CdR low;5:5-azaCdR mid;6:5-aza-CdR high;7:As2O3 + 5-aza-CdR low;8:As2O3 + 5-azaCdR mid;9:As2O3 + 5-aza-CdR high
图2 JAK3基因在U937细胞中的表达
Figure 2 JAK3 mRNA expression in U937 cells |
|
|
表 3 单用As2O3 及联合用药处理后JAK3基因在U937细胞中的表达比较 Table 3 Comparison of JAK3 gene expression in U937 cells treated with As2O3 and the combination |
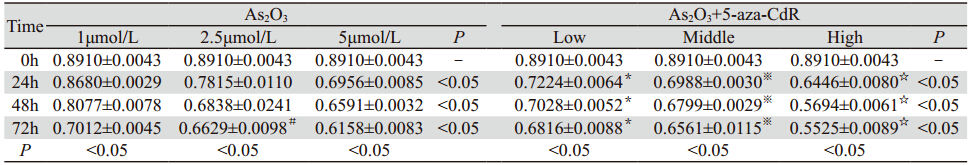
随着药物浓度的增加和作用时间的延长, JAK3的表达水平下降,与未用药的对照组比 较差异均有统计学意义(P<0.05)。除低浓度 (0.5 μmol/L)时,空白组和24 h差异无统计学 意义(P=0.052),48和72 h差异无统计学意义 (P=0.235);中浓度(1 μmol/L),48和72 h差异无统计学意义(P=0.120)外,其余各浓度在各 时段的表达变化经两两比较差异均有统计学意义 (P<0.05)。在三个时段(24、48、72 h)内, 低、中、高浓度间的表达变化经两两比较,差异 均有统计学意义(P<0.05),见图 2、表 4。
|
|
表 4 单用5-aza-CdR及联合用药处理后JAK3基因在U937细胞中的表达 Table 4 Comparison of JAK3 gene expression in U937 cells treated with 5-aza-CdR and the combination |

随着药物浓度的增加和作用时间的延长, JAK3的表达水平下降,与未用药的对照组以 及各组之间两两比较,差异均有统计学意义 (P<0.05),合用药均比单用药时的表达水平下 降,差异有统计学意义(P<0.05)。 2.3 TYK2基因在U937细胞中的表达
单独应用As2O3 和5- aza- CdR时以及两药合 用时,随着药物浓度的增加和作用时间的延 长,TYK2的表达水平降低,与未用药的对照 组,以及各组间两两比较差异均有统计学意义 (P<0.05)。合用药均比单独用药时的表达水平 降低,差异有统计学意义(P<0.05),见图 3,表 5、6。
 |
1:As2O3 low;2:As2O3 mid;3:3:As2O3 high;4:5-aza-CdR low;5:5-azaCdR mid;6:5-aza-CdR high;7:As2O3 + 5-aza-CdR low;8:As2O3 + 5-azaCdR mid;9:As2O3 + 5-aza-CdR high
图3 TYK2基因在U937细胞中的表达
Figure 3 TYK2 mRNA expression in U937 cells |
|
|
表 5 单用As2O3 及联合用药处理后JAK3基因在U937细胞中的表达比较 Table 5 Comparison of TYK2 gene expressions in U937 cells after As2O3 alone and the combined treatment |
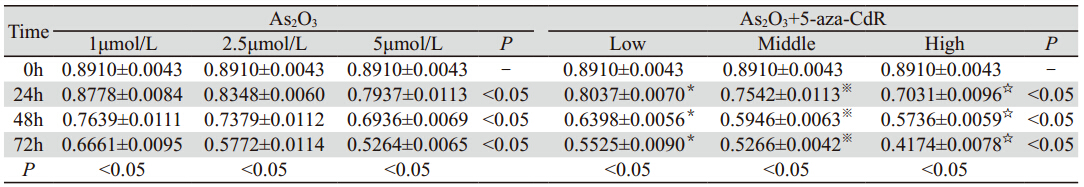
|
|
表 6 单用5-aza-CdR及联合用药处理后TYK2基因在U937细胞中的表达的比较 Table 6 Comparison of TYK2 gene expressions in U937 cells treated with 5-aza-CdR alone and the combination |

通过对As2O3 和5-aza-CdR作用于U937细胞株 后基因表达做直线回归分析后,可以看出,直线 回归方程均有意义(P=0),JAK3和TYK2的基因 表达量同药物作用时间及浓度呈线性相关,呈负 相关,随着作用时间的延长和药物浓度的增加, JAK3和TYK2的基因表达量下降(回归系数b<0)。 在三种浓度分别作用下,JAK3的下降程度均大于 TYK2的下降程度(|bJAK3|>|bTYK2|),但在三 个药物作用时间段中,仅48 h时TYK2的下降程度 大于JAK3的下降程度(|bTYK2|>|bJAK3|),见表 7、8。
|
|
表 7 相同浓度下TYK2,JAK3基因在U937细胞中的表达变化的回归系数比较 Table 7 Comparison of regression coefficients of TYK2, JAK3 expression changes in U937 cells under the same concentration of drugs |

|
|
表 8 相同处理时间后TYK2,JAK3基因在U937细胞中的表达变化的回归系数比较 Table 8 Comparison of regression coefficients of TYK2, JAK3 expression changes in U937 cells with the same treatment time |

基因启动子区域CpG岛的高度甲基化是白血 病(leukemia)等血液系统恶性肿瘤的一种发病机 制,大概有90%的血液系统恶性肿瘤都会至少有 一个基因高度甲基化。DNA高度甲基化成为髓系 白血病的重要致病机制[4]。SHP-1主要表达于造血 细胞,被称为造血细胞磷酸酶(hematopoietic cell phosphatase,HCP),在信号转导中主要起负调 控作用,抑制细胞增殖[5]与肿瘤的形成及生长呈负 相关。韩颖等[6]研究发现,SHP-1的低表达或不表 达与AL的发病密切相关。JAK [7]是一种蛋白酪氨 酸激酶(PTK),迄今为止共发现4种家族成员: JAK1、JAK2、JAK3和TYK2。除了JAK3外,其 余3种成员广泛分布于多种组织细胞中。而JAK3却 仅局限于白细胞中,在活化的T细胞、B细胞和单 核细胞中有高水平的表达[8]。SHP-1主要通过去除 JAKs蛋白磷酸化对JAKs进行调解,也可加速JAKs 蛋白的降解而下调JAK/STAT途径[9]。
5-aza-CdR是一种甲基化抑制剂,苏庸春等[10] 研究发现5-aza-CdR作用于肿瘤细胞的主要机制是 通过其去甲基化作用,重新激活了那些由于DNA 过度甲基化而受到抑制的抑癌基因或多种细胞调控 基因,从而使细胞向终末分化、凋亡或衰老,并促 进细胞凋亡、抑制细胞增殖。As2O3 主要用于急性 早幼粒细胞白血病(APL)、骨髓增生异常综合征 等恶性血液病的诱导分化治疗,取得较好疗效。沈 松菲等[11]研究发现As2O3 可诱导Jurkat细胞中异常甲 基化的hdpr1基因去甲基化并使其恢复表达。
本实验采用实时定量PCR技术,检测到白血 病细胞株U937中SHP-1基因低表达,JAK3、TYK2 高表达。在应用了甲基化抑制剂5-aza-CdR与As2O3 后,U937细胞的增殖受到不同程度的抑制,有 剂量和时间依赖性。单独应用这两种药物后,都 可以使原来低表达的SHP-1表达升高,并且随着 药物浓度的增高和作用时间的延长其表达量也增 加,而JAK3、TYK2基因表达也随之下降,说明 基因的表达水平与药物的作用时间和浓度有关。 联合应用这两种药物,发现SHP-1基因表达水平的 升高和JAK3、TYK2基因表达水平的下降幅度, 比单独应用时更为显著。杨琳等[12]通过对63例成 人急性髓系白血病患者的SHP-1及JAKs基因表达 研究发现,在mRNA水平,SHP-1表达与JAK1、 JAK3呈负相关。故推测,SHP-1基因的重新表达 与它发生去甲基化有关系,对JAK/STAT通路发挥了的负调控作用,使JAK3、TYK2基因的表达水 平下降。较长的作用时间,较高的药物浓度以及 两种药物联合后均可以加强其去甲基化的作用。 在JAK/STAT通路中,可能适合的浓度更能加强 SHP-1对JAK3的负调控作用。
综上所述,异常激活JAK/STAT信号转导通 路,可导致白血病的发生。JAK3、TYK2基因 的表达上调,可以正性激活JAK/STAT信号转导 通路。JAK3、TYK2、SHP-1都参与了白血病的 发病。SHP-1在白血病的发病中起着负性调节作 用。JAK3、TYK2在白血病的发病中起着正性调 节作用,它的高表达促进白血病的发生。As2O3 和 5-aza-CdR对U937细胞株中抑癌基因的去甲基化作 用为我们提供了用去甲基化药物治疗肿瘤的一种 新思路。同时,SHP-1基因的重新表达、JAK3、 TYK2的降低表达,进一步说明了SHP-1在白血病 中发挥着重要的负调节功能。
| [1] | Witkiewicz A, Raghunath P, Wasik A, et al. Loss of SHP-1 tyrosine phosphatase expression correlates with the advanced stages of cutaneous T-cell lymphoma[J]. Ham Pathol,2007,38(3):462-7. |
| [2] | Luo JM, Li Y, Yang L,et al. Effect of a methylation inhibitor 5-aza-2’-deoxycytidine on SHP-1 gene expression, proliferation and apoptosis in K562 cells[J]. Zhongguo Shi Yan Xue Ye Xue Za Zhi,2009,17(2):309-14.[罗建民,李燕,杨琳,等.甲基化抑制剂 5-aza-2′-deoxycytidine对K562细胞SHP-1基因表达及细胞增殖 与凋亡的影响[J]. 中国实验血液学杂志,2009,17(2):309-14.] |
| [3] | Ma ZJ,Zhang WJ,Zhao PR,et al. Demethylation of estrogen r ecep to r- α in b r eas t can cer cells MDA- MB- 2 3 1 af ter treated by arsenic trioxide[J]. Zhong Liu Fang Zhi Yan Jiu,2011,38(7):749-51. [马志俊,张伟杰,赵培荣,等. 三氧化二砷对乳腺癌细胞MDA-MB-231雌激素受体α的去甲基化作用[J]. 肿瘤防治研究,2011,38(7):749-51.] |
| [4] | Leo E, Mancini M, Aluigi M,et al. DNA hypermethylation promotes the low expression of pro-apoptotic BCL2L11 associated with BCR-ABL1 fusion gene of chronic myeloid leukaemia[J]. Br J Haematol,2012,159(3):373-6. |
| [5] | Mauldin IS, Tung KS, Lorenz UM. The tyrosine phosphatase SHP-1 dampens murine Th17 development[J]. Blood,2012, 119(19):4419-29. |
| [6] | Han Y, Luo JM, Jia XH,et al. Expression and clinical value of SHP-1 and c-kit in acute leukemia[J]. Zhongguo Shi Yan Xue Ye Xue Za Zhi, 2006,14(5):867-71. [韩颖,罗建民,贾晓辉,等. 急性 白血病患者SHP-1与c-kit基因表达及临床意义[J]. 中国实验血液学杂志,2006,14(5):867-71.] |
| [7] | Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty[J]. Immunity,2012,36(4):503-14. |
| [8] | Jaime-Figueroa S,De Vicente J,Hermann J,et al. Discovery of a series of novel 5H-pyrrolo[2,3-b]pyrazine-2-phenyl ethers, as potent JAK3 kinase inhibitors[J]. Bioorg Med Chem Lett,2013,23(9):2522-6. |
| [9] | Alicea-Velázquez NL, Jakoncic J, Boggon TJ. Structure-guided studies of the SHP-1/JAK1 interaction provide new insights into phosphatase catalytic domain substrate recognition[J]. J Struct Biol,2013,181(3):243-51. |
| [10] | Su YC,Xu HZ,Yu J, et al. Effects of 5-azacytidine on methylation state of P16, DAPK and MGMT genes in HL60 cells[J]. Chongqin Yi Ke Da Xue Xue Bao,2012,37(10):864-7. [苏庸春,徐红珍,于洁, 等. 5-氮杂胞苷对HL60细胞P16、DAPK及MGMT基因甲基化状态影响[J] .重庆医科大学学报,2012,37(10):864-7.] |
| [11] | Shen SF, Shen JZ, Fu HY,et al. Mechanism of As203 on hdpr1 gene demethylation in Jurkat cell line[J]. Zhongguo Shi Yan Xue Ye Xue Za Zhi,2010,18(6):1484-8. [沈松菲,沈建箴,付海英,等. As203对Jurkat细胞株hdpr1基因去甲基化作用机制的研究[J]. 中国实验血液学杂志,2010,18(6):1484-8.] |
| [12] | Yang L, Luo JM, Wen SP, et al. Expressions of SHP-1 and JAKs genes in AML patients and their correlation[J]. Zhong Liu Fang Zhi Yan Jiu,2009,36(12):1031-4.[杨琳,罗建民,温树鹏,等. SHP-1 和JAKs基因在成人急性髓系白血病患者中的异常表达及其相关性[J].肿瘤防治研究,2009,36(12):1031-4.] |