 2014, Vol.41
2014, Vol.41


外源性AKT1增强上皮性卵巢癌细胞侵袭和迁移能力的机制
引用本文
张海燕,孙 红. 外源性AKT1增强上皮性卵巢癌细胞侵袭和迁移能力的机制[J]. 肿瘤防治研究, 2014, 41(06): 557-561. 
ZENGZHANG Haiyan, SUN Hong. Mechanism of AKT1 Enhancing Migration and Invasion of Epithelial Ovarian Cancer
Cells. Cancer Research on Prevention and Treatment, 2014, 41(06): 557-561.
外源性AKT1增强上皮性卵巢癌细胞侵袭和迁移能力的机制
200011 上海,复旦大学附属妇产科医院妇科
摘要:目的 探讨AKT1基因对上皮性卵巢癌SKOV3细胞迁移和侵袭能力的影响及相关分子机制。方法 针对AKT1基因,设计并构建shRNA质粒和真核表达质粒,双向调节SKOV3细胞中AKT1的表
达,运用RT-PCR和western blot检测转染效率。运用Wound healing和Transwell-Matrigel方法检测转染前
后细胞迁移和侵袭能力的变化。RT-PCR法检测与细胞运动侵袭相关分子CXCR4、VEGF、MMP-2、
MMP-9和uPA在mRNA水平的表达变化。结果 成功构建AKT1基因的真核表达质粒pEF-1α-AKT1和
靶向抑制AKT1基因的shRNA表达质粒pRNAT-AKT1。转染上皮性卵巢癌SKOV3细胞后,能有效调控
p-AKT表达。参照未转染组和空载体转染组,外源性AKT1促进细胞迁移和侵袭,CXCR4、VEGF、
MMP-2和uPA的mRNA表达水平升高。shRNA靶向抑制AKT1基因的表达可抑制细胞迁移和侵袭,
CXCR4、VEGF、MMP-2和uPA的mRNA表达水平下降。结论 AKT1可能通过调控CXCR4、VEGF、
MMP-2和uPA的转录水平来影响细胞侵袭和运动能力。
关键词:
上皮性卵巢癌
AKT1
信号通路
Mechanism of AKT1 Enhancing Migration and Invasion of Epithelial Ovarian Cancer Cells
Department of Gynecology,Hospital of Obstetrics and Gynecology,Affiliated of Fudan
University,Shanghai 200011,China
Abstract:Objective To explore the effects of AKT1 gene on the invasion and migration of epithelial
ovarian cancer cells SKOV3 and its mechanism. Methods Recombinant plasmid carrying the full-length
AKT1 cDNA and AKT1 shRNA plasmid were constructed. Plasmids were transfected into SKOV3 cells to
dual-directionally regulate AKT1 expression. RT-PCR and western blot were used to detect the transfection
efficiency. Wound healing assay was used to analyze cell migration. Transwell-Matrigel assay was used
to analyze cell invasion. RT-PCR was used to explore the expression of CXCR4, VEGF, MMP-2, MMP-9
and uPA which were related with cell invasion and migration at mRNA level. Results A plasmid (pEF-
1α-AKT1) containing full length of AKT1 cDNA and a specifi c AKT1-targeted shRNA expression plasmid
were constructed successfully, which could effectively regulate the activation of p-AKT after SKOV3 cells
transfected. Compared with untranfected cells or non-target shRNA transfected cells, up-regulation AKT1
promoted cell migration and invasion, and increased the expression of CXCR4, VEGF, MMP-2, and uPA at
mRNA level(P<0.05). On the other hand, down-regulation AKT1 inhibited cell migration and invasion, and
decreased the expression of CXCR4, VEGF, MMP-2, and uPA at mRNA level(P<0.05). Conclusion AKT1
might impact cell migration and invasion by regulating the expression of CXCR4, VEGF, MMP-2, and uPA at
mRNA level.
Key words:
Epithelial ovarian cancer
AKT1
Signal pathway
0 引言
2.2 AKT基因对SKOV3细胞侵袭能力的影响
2.3 AKT基因对SKOV3细胞迁移能力的影响
2.4 RT-PCR检测MMP-2、MMP-9、uPA、VEGF
和CXCR4 mRNA的表达水平
3 讨论
AKT在正常卵巢组织和癌组织中激活水平的 显著差异提示其在肿瘤发生发展中的重要作用。 本文前期研究发现,上皮性卵巢癌中AKT异常激 活不仅是上游PI3K分子异常激活的后续效应, 同时与AKT基因自身扩增密切相关。既往研究表 明,AKT异常激活可促进卵巢癌细胞增殖,抑制细胞凋亡。但AKT异常激活对卵巢癌细胞的运动 和侵袭的影响及相关机制,仍有待深入探讨。本 研究中,构建了外源性AKT1基因真核表达质粒和 靶向AKT1基因的shRNA质粒,采用脂质体介导的 方法,分别转染SKOV3细胞,观察细胞转染前后 p-AKT表达水平的变化,检测其对细胞侵袭和迁 移等生物学行为的影响及机制。 1 材料与方法 1.1 材料
上皮性卵巢癌细胞株SKOV3购自ATCC。鼠 抗人磷酸化AKT(Ser473)单克隆抗体、兔抗人 AKT多克隆抗体、兔、鼠抗人GAPDH单克隆抗体 均购自美国Cell Signal公司。TRIZOL、RT-PCR试 剂盒购自美国Invitrogen公司。培养液及胎牛血清 购自美国GIBICO公司。RT-PCR试剂及限制性内 切酶盒购自Fermentas公司。质粒抽提试剂盒购自 美国QIAGEN公司。Lipofectamine™2000购自美国 Invitrogene公司。 1.2 构建pEF6/Myc-AKT1质粒
根据GeneBank中AKT1基因序列(收录号: NM0332300),应用Primer5.0软件设计引物, 在5’端和3’端引物加上BamHⅠ、 XbaⅠ酶切位 点。扩增AKT1反义核酸引物序列为:上游引 物:5’-GGA TCC ATG AAG ACG GAG CGG CCC CG-3’,下游引物:5’-TCT AGA TCA GGC CGT CGC GCT GGG CG-3。以SKOV3 cDNA模板进行 高保真PCR扩增,扩增片段1 443 bp。产物经酶 切、回收、纯化后与载体片段 pEF6/Myc-His A酶 切片段相连,再次经酶切、回收、纯化后转化感 受态体菌DH5α。摇菌后进行质粒抽提、鉴定,获 得导入AKT1基因全长cDNA的质粒。 1.3 构建短发卡式RNA-AKT1质粒(shRNAAKT1)
参照siRNA的设计原则,在 AKT1(序列号: NM033230)的编码序列中,设计19~21 nt的DNA片 段,并利用BLAST进行查询,确定其为特异性序 列。阴性对照选取国际通用的non-target siRNA。 根据选定的siRNA序列合成两条模板单链,每条单 链均包括该基因的正义链和反义链,中间以9个脱 氧核苷酸的Loop结构相连,后面续接RNA PollyⅢ 聚合酶转录终止位点,同时模板链两端分别设计 BamH Ⅰ和Hind Ⅲ的内切酶位点。产物经酶切、 回收、纯化后与双酶切的线化pRNA质粒载体相 连,再次经酶切、回收、纯化后转化感受态体菌 DH5α。摇菌后进行质粒抽提、鉴定,-20℃保存 待用。 1.4 重组质粒转染SKOV3细胞
SKOV3细胞以2.5×106/孔的密度接种至6孔 板,待细胞汇聚率达到90%左右,按照推荐剂量 准备质粒-LipofectamineTM2000复合物,加入到培 养孔中,分为实验组(含目的基因质粒转染组)、阴 性对照组(空质粒转染组或含non-target shRNA转染 组)、空白对照组(未转染组)。在转染后6 h改换含 10%胎牛血清的DMEM高糖培养液。 1.5 Transwell小室检测细胞的侵袭能力
Matrigel(50 mg/L)按1:8稀释,包被Transwell 小室底部,4℃过夜;上室加入200 μl含0.5%BSA 的DMEM培养液,水化基底膜,37℃ 30 min,加 入含5×103个细胞悬液200 μl;下室加入800 μl 5% FBS培养液;在37℃、5%CO2培养箱培养24 h; PBS淋洗,并用棉签擦去微孔膜上层的细胞,95% 乙醇室温固定20 min后,苏木精染色10 min,1% 盐酸酒精分化,自来水淋洗;电子显微镜观察、 拍照、计数5个高倍视野(×200)下穿过滤膜的细 胞数,取其平均值。各组细胞穿过Transwell的数 量作为比较细胞侵袭能力的指标。实验重复3次。 1.6 划痕试验检测细胞的迁移能力
在12孔板接种细胞5×105/孔,设置3复孔;待 细胞密度达到90%时,用10 μl的枪头做垂直划痕; PBS洗涤3次以去除细胞碎片;加入含5% FBS的培 养基,在37℃、5%CO2培养箱培养,不同时间点 (0,24,48h)显微镜下观察细胞向划痕区的生 长情况并拍照,测量各组细胞任意三个部位的不 同伤口的宽度,相对运动率=[W(t=0 h)-W(t=24 h,48 h)]/W(t=0 h),计数三次实验的平均值,作为 评价细胞迁移能力的指标。 1.7 Western blot分析总AKT和磷酸化AKT的蛋白 表达水平
细胞经无血清培养24 h同步化后,加入含10% 血清的培养液刺激2 h后,弃去上清液,PBS淋 洗,甩干,RIPA提取总蛋白并BCA法测定蛋白水 平。SDS聚丙烯酰胺凝胶电泳后,稳定电流270 mA在4℃90 min转移到硝酸纤维素膜上,放入5% 封闭液中,室温摇床振荡2 h,单克隆鼠抗人磷 酸化AKT (Ser473)(1:500),或单克隆兔抗人AKT (1:1 000)以及内参GAPDH (1:5 000)的封闭液,赶 尽气泡,用封口机封口,4℃过夜。PBST轻轻漂 洗15 min×3次;将膜转入另一杂交袋中,加入荧 光二抗,室温避光孵育1 h (1:3000),应用LI-COR Odyssey Infrared Imaging System扫描检测。得出的 IOD值经内参GAPDH校正后进行统计学分析。 1.8 RT-PCR分析AKT激活对MMP-2、MMP-9、 uPA、CXCR4和VEGF mRNA表达水平的影响
依据试剂盒步骤进行提取总RNA。引物由上 海生工合成。AKT1上游引物为5’-TCT ATG GCG CTG AGA TTG TG-3’,下游引物为5’- CTT AAT GTG CCC GTC CTT GT-3’(合成片段 112bp,退 火温度59℃);VEGF上游引物为5’-GC TCT ACC TCC ACC ATG CCA-3’,下游引物为5’-AGC TCA TCT CTC CTA TGT GC-3’(合成片段 320bp,退 火温度51.5℃);CXCR4上游引物为5’-TCT CGG TCC ACT CTT GTT-3’,下游引物为5’-GCC ACT CAG GAG GAT TAC-3’(合成片段362bp,退火温度 60℃);MMP-2上游引物为5’-CAT AGG ATG TGC CCT GGA AG-3’,下游引物为5’-TGG TCC TGG TTG TAG AAG GG-3’(合成片段 427bp,退火温 度58.5℃)。MMP-9上游引物为5’- GGA TGG GAA GTA CTG GCG ATT-3’,下游引物为5’-CAC TTG GTC CAC CTG GTT CAA-3’(合成片段543bp,退 火温度59℃)。uPA上游引物为5’-CGG AAT TCC AGC AAC GAA CTT-3’,下游引物为5’-GAT CCG CTA GCT TTC AGT CTG-3’(合成片段376bp,退火 温度55℃)。GAPDH上游引物为5’-GAA GGT GAA GGT CGG AGT C-3’,下游引物为5’- GAA GAT GGT GAT GGG ATT TC-3’ (合成片段 220bp,退火 温度50~60℃)。各样本PCR产物通过1.2%琼脂糖凝 胶电泳后,应用凝胶成像扫描及分析系统,用各 样本的目的基因条带灰度值与相应GAPDH条带灰 度值相比,求其比值。 1.9 统计学方法
应用SPSS11.5软件进行统计分析,实验数据
以均数±标准差(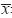 ±s)表示。用Excel软件作图,
采用单因素方差分析及χ2检验,P<0.05为有差异
具有统计学意义。
2 结果
2.1 重组质粒对上皮性卵巢癌SKOV3细胞中
AKT1 mRNA表达和蛋白激活水平的调节
±s)表示。用Excel软件作图,
采用单因素方差分析及χ2检验,P<0.05为有差异
具有统计学意义。
2 结果
2.1 重组质粒对上皮性卵巢癌SKOV3细胞中
AKT1 mRNA表达和蛋白激活水平的调节
RT-PCR法检测转染前后AKT1 mRNA表达水 平的变化,分别以未转染组细胞和阴性对照组细 胞为对照。外源性pEF-1α-AKT可明显上调AKT1 mRNA转录水平;而shRNA表达质粒可显著抑制 AKT1mRNA表达,差异有统计学意义(P=0.029; P=0.032),见图 1A。Western blot检测转染前 后AKT总蛋白和磷酸化AKT表达水平的变化与 mRNA变化水平是一致的,见图 1B。
 |
A:RT-PCR determined the transfective effi ciency in SKOV3 cells on AKT1 mRNA level;B:Western blot determined the transfective effi ciency in SKOV3 cells by detecting the activity of AKT;1: SKOV3 cells;2:SKOV3/Negative cells;3:SKOV3/pRNAT-AKT cells;4:SKOV3 cells;5:SKOV3/pEF- 1α cells;6:SKOV3/pEF-1α-AKT cells;*:P=0.029,P=0.032,compared with control group and the negative control group,respectively,n=3 in each group;**:P=0.0065,P=0.0049 compared with control group and negative control group,respectively,n=3 in each group; take GAPDH as control 图 1 RT-PCR、Western blot检测质粒转染前后SKOV3细胞中AKT1 mRNA、AKT总蛋白和磷酸化AKT表达水平 Figure 1 RT-PCR and Western blot were used to determine transfective effi ciency in SKOV3 cells by detecting the expression of AKT mRNA level and activity of AKT respectively |
Transwell小室培养24 h,上调AKT基因后 SKOV3/pEF1-α-AKT细胞穿透Matrigel包被膜的 细胞数(132.92±7.32)显著多于SKOV3/pEF1-α 细胞(60.34±5.23)和未转染SKOV3细胞(57.97± 6.73)的穿透数,差异均有统计学意义(P=0.037, P=0.034),见图 2A。相反,下调AKT1基因 Transwell小室培养24h,SKOV3/pRNAT-AKT1细胞 穿透Matrigel包被膜的细胞数(20.21±3.89)显著少 于阴性对照SKOV3/Negative细胞(62.34±4.23)和 未转染SKOV3细胞(57.97±6.73)的穿透数,均有统 计学意义(P=0.018;P=0.025),见图 2B。
 |
A:Transwell assay showed upregulation AKT enhanced SKOV3 cells’ invasion;B:Transwell assay showed AKT siRNA inhibited SKOV3 cells’ invasion(hematoxylin-staining ×200,A *:P=0.037,P=0.034,compared with the control group and the negative control group,respectively,n=3 in each group;B*:P=0.018; P=0.025,compared with the control group and the negative control group,respectively,n=3 in each group 图 2 Transwell小室检测质粒转染前后对SKOV3细胞侵袭能力的影响 Figure 2 Transwell assay detected the effect of AKT regulation on the invasion of SKOV3 cells |
划痕后培养24 h,相对于未转染质的空白细胞 组和和阴性对照细胞组,SKOV3/pEF1-α-AKT细 胞明显向划痕处迁移;划痕48 h,SKOV3/pEF1-α- AKT细胞的划痕基本修复,见图 3A。外源性AKT 可促进SKOV3细胞的迁移。反之,在划痕培养48 h时,SKOV3/pRNAT-AKT细胞的划痕明显宽于未 转染SKOV3细胞和SKOV3/Negative细胞的划痕, 从而提示AKT1基因干扰抑制SKOV3细胞迁移,见 图 3。
 |
A:wound healing assay showed upregulation AKT increased cell migration rate in SKOV3 cells;B:wound healing assay showed downregulation AKT decreased cell migration rate in SKOV3 cells. A**:P=0.0061,P=0.0078,compared with the control group and the negative control group,respectively, n=3 in each group;B**:P=0.0053,P=0.0058,compared with the control group and the negative control group,respectively,n=3 in each group 图 3 划痕实验检测AKT基因对SKOV3细胞迁移能力的影响 Figure 3 Wound healing assay detected the effect of AKT regulation on the migration rate of SKOV3 cells |
为探讨可能的调控机制,RT-PCR分析侵袭 及迁移相关分子变化。上调AKT可促进MMP-2、 uPA、VEGF和CXCR4 mRNA表达,反之下调AKT 则抑制MMP-2、uPA、VEGF和CXCR4 mRNA表 达水平。但对MMP-9 mRNA表达水平的影响不明 显,见图 4。
 |
1:SKOV3 cells;2:SKOV3/pEF-1α cells;3: SKOV3/pEF-1α-AKT cells;4:SKOV3 cell;5: SKOV3/Negative cells;6:SKOV3/pRNATAKT cells;MMP-2matrix metalloproteinase-2 MMP-9 matrix metalloproteinase-9uPA human urokinase plasminogen activator VEGF vascular endothelial growth factor CXCR4 chemokine (C-X-C motif) receptor 4 图 4 RT-PCR检测AKT基因对SKOV3 细胞中MMP-2、MMP-9、uPA、VEGF 和CXCR4 mRNA表达水平的影响 Figure 4 RT-PCR analysis determined the transfective efficiency in SKOV3 cells and changes of CXCR4,VEGF, MMP-2,MMP-9 and uPA at mRNA level |
本文通过构建AKT1表达质粒和AKT shRNA 调控AKT的mRNA水平,发现相同培养条件下, 相对于空白细胞组和阴性对照组,转染细胞组的 AKT磷酸化水平发生了相应的调节。
肿瘤细胞的侵袭和迁移能力是肿瘤细胞转移 潜能的体现。Transwell-Matrigel法是用来检测细胞 侵袭的量化分析模型。划痕实验粗略反映了细胞 的迁移能力。本研究显示,shRNA干扰细胞后, 细胞侵袭和迁移能力下降超过50%;反之,外源性 AKT1转染SKOV3细胞后,细胞侵袭和迁移能力增 强,从而说明AKT基因在卵巢癌中可调控细胞的 侵袭和迁移能力,AKT基因磷酸化水平越高,则 细胞越容易浸润周围组织乃至远处转移。
至于AKT基因对癌细胞侵袭和转移的调控机 制,以往研究发现,PI3K/AKT信号通路可通过激 活mTOR以及其下游核糖体蛋白p70S6K,促进肌 动蛋白的细丝重构,促进细胞运动[1];AKT可上调 NF-κB的转录活性,增加肿瘤细胞的引动功能,有 助于癌细胞侵袭[2];PI3K/AKT信号通路可上调基 质金属蛋白酶-2(MMP-2)[3]、MMP-9[4]mRNA和 蛋白质表达,降解细胞外基质促进肿瘤细胞的转 移。 本研究采用RT-PCR法检测了一系列与侵袭或 转移相关分子的mRNA水平变化,研究结果与以 往的研究结果有不同之处。
MMPs和uPA被认定是对肿瘤细胞本身的增 殖和生长所必不可少的,同时也是与肿瘤侵袭转 移密切相关的两套系统。本研究显示,转染外 源性AKT1的SKOV3细胞比阴性对照组细胞表达 MMP-2和uPA的水平明显升高,但MMP-9未见显 著增加;反之,shRNA靶向抑制AKT1基因显著 下调MMP-2和uPA的表达水平,但MMP-9未见显 著改变,提示AKT1基因可能通过调控MMP-2和 uPA,影响其对基质的降解,从而调控肿瘤的侵袭 和转移。尽管本研究显示,AKT对MMP-9的表达 影响不明显,但不排除组织特异性的差异,今后 将通过检测多种卵巢癌细胞株来进一步证实。
CXCR4是趋化因子SDF-1α配体[5]。Hashimoto 等[6]研究认为SDF-1α/CXCR4轴异常是导致胃癌 腹腔转移机制之一,阻断CXCR4不仅抑制细胞 转移,同时抑制mTOR激活,诱导细胞自噬。本 研究发现,AKT1基因亦可能通过调控VEGF、 CXCR4,影响各自信号传递,从而控制卵巢癌细 胞的侵袭和转移,但具体的调控网络仍需进一步 深入。
综上,AKT信号通路可能通过调控MMP-2、 uPA、CXCR4、VEGF转录参与上皮性卵巢癌细胞 的侵袭和转移。因此,靶向AKT可深入探讨恶性 肿瘤的表型调控机制,为肿瘤治疗筛选有效靶位。
参考文献
| [1] | Winbanks CE,Grimwood L,Gasser A, et al. Role of the phosphatidylinositol 3-kinase and mTOR pathways in the regulation of renal fi broblast function and differentiation[J]. Int J Biochem Cell Biol,2007,39(1):206-19. |
| [2] | Shih YW, Chen PS, Wu CH, et al. Alpha-chaconine-reduced metastasis involves a PI3K/Akt signaling pathway with downregulation of NF-kappaB in human lung adenocarcinoma A549 cells[J]. J Agric Food Chem,2007,55(26):11035-43. |
| [3] | Lee SJ,Seo KW,Yun MR, et al. 4-Hydroxynonenal enhances MMP-2 production in vascular smooth muscle cells via mitochondrial ROS-mediated activation of the Akt/NF-kappaB signaling pathways[J]. Free Radic Med,2008,45(10):1487-92. |
| [4] | Chen JS,Wang Q,Fu XH, et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: association with MMP-9[J].Hepatol Res,2009,39(2):177-86. |
| [5] | Wong D, Korz W. Translating an antagonist of chemokine receptor CXCR4: from bench to bedside[J]. Clin Cancer Res,2008,14(24):7975-80. |
| [6] | Hashimoto I, Koizumi K, Tatematsu M, et al. Blocking on the CXCR4/mTOR signalling pathway induces the anti-metastatic properties and autophagic cell death in peritoneal disseminated gastric cancer cells[J]. Eur J Cancer,2008,44(7):1022-9. |