
-
摘要:目的
探讨铜死亡相关基因与肝癌生存预后的关系。
方法通过收集TCGA数据库中肝癌患者的临床信息和相应的RNA-seq数据,分析10个铜死亡相关基因在肝癌组织和正常组织中的差异表达。进一步使用一致性聚类以确定新的肝癌亚型,比较两个亚型之间总体生存率和临床特征的差异。单因素Cox回归分析筛选与预后相关的铜死亡基因,并利用LASSO回归分析构建风险模型。
结果与正常组织相比,肝癌组织中FDX1表现下调,其余9个基因表现上调。聚类分析示,Cluster1的预后较差。基于单因素Cox回归分析和LASSO回归分析筛选出5个预后相关基因(LIPT1、DLAT、MTF1、GLS、CDKN2A)并构建风险模型。与其他临床特征相比,该预后模型的风险评分被确认为独立的预后因素。
结论通过生物信息学分析构建了5个铜死亡相关基因的肝癌预后模型,有可能作为肿瘤诊断分子标志物和潜在的治疗靶点。
Abstract:ObjectiveTo explore the relationship of cuprotosis-related genes with survival rate and prognosis in patients with liver cancer.
MethodsBy collecting clinical information and corresponding RNA-seq data of patients with liver cancer in the TCGA database, the differential expression levels of 10 cuprotosis-related genes in liver cancer and normal tissues was analyzed. Novel liver cancer subtypes were identified through consistent clustering, and differences in overall survival and clinicopathological factors were compared between the two subtypes. Univariate Cox regression analysis was used in screening cuprotosis genes associated with prognosis, and LASSO regression analysis was used in constructing a risk model.
ResultsFDX1 was down-regulated, and the other nine genes were up-regulated in HCC tissues compared with normal tissues. Cluster analysis showed that the prognosis of Cluster1 was poor. Five prognostic genes (LIPT1, DLAT, MTF1, GLS, and CDKN2A) were screened out through univariate Cox regression analysis and LASSO regression analysis for risk model construction. The risk score of this prognostic model was identified as an independent prognostic factor compared with other clinical features.
ConclusionThrough bioinformatics analysis, a liver cancer prognosis model of five cuprotosis-related genes is constructed, which may be used as molecular markers for tumor diagnosis and are potential therapeutic targets.
-
Key words:
- Bioinformatics /
- Liver cancer /
- Cuprotosis /
- Prognostic model
-
0 引言
肝癌是全球最常见的恶性肿瘤之一,发病率全球排名第五,男性死亡率排名第二[1]。尽管早期肝癌患者可行肿瘤切除、肝移植等手术治疗,但由于供体短缺等原因,大多数患者仍不适用。很多患者被诊断为疾病晚期,因此错过了接受外科手术等根治性治疗的机会。此外,对于接受手术切除的患者,复发仍然是一个主要问题,而且50%复发的患者在一年内死亡[2-3]。由于复杂的病因及高复发性使肝癌预后预测具有挑战性,考虑到肝癌治疗策略有限,因此还需要开发新的预后模型。
随着微阵列技术和高通量测序技术的发展,人们现在能够通过生物信息学分析来识别与肿瘤预后和进展相关的关键基因,并在此基础上发现了许多预后标志物,建立了各种预后模型[4-5]。铜离子参与体内众多生化反应,当体内铜离子过高时,会对细胞产生毒性,改变氧化还原状态。有研究表明,与健康人群相比,癌症患者的血清铜水平升高,并与疾病的严重程度和对治疗的反应相关[6]。最近,一种不同于已知细胞死亡机制的新型细胞死亡方式引起人们广泛关注,它主要通过铜离子与线粒体中的三羧酸循环中的脂酰化成分直接结合,从而导致脂酰化蛋白质聚集和随后的铁硫簇蛋白下调,最终使得蛋白质毒性应激并导致细胞死亡[7-8],并且从中发现了几个与铜死亡相关的基因,这可能为预测肝癌患者的预后提供新的策略。
本研究采用生物信息学的方法分析铜死亡相关基因在肝癌与正常组织中的差异表达,利用聚类分析和LASSO回归分析筛选出与预后相关的基因并构建风险预测模型,以期为肝癌患者早期诊断和预后评估提供参考依据。
1 资料与方法
1.1 数据集的获取与处理
从肿瘤基因组图谱(TCGA)数据库(https://portal.gdc.cancer.gov/)下载了371例肝癌患者肿瘤组织和50例正常肝组织的RNA测序(RNA-seq)数据集和临床资料。采用R软件“Limma”包对转录组基因表达谱进行预处理。临床资料主要包括患者的年龄、性别、肿瘤分级、肿瘤分期、生存时间和生存状态等信息。此外,为了减少该分析中潜在的统计偏差,排除了生存时间未知且无生存状态的患者。
1.2 铜死亡相关基因的获取及差异分析
选择目前已知的10个铜死亡相关基因,提取这10个基因的表达矩阵,并使用Wilcoxon秩和检验进行组间差异表达分析。利用“corrplot”软件包进行相关性分析,基于“pheatmap”软件包绘制热图。
1.3 聚类分析
移除50例正常组织样本,使用“Consensus-ClusterPlus”软件包对肝癌患者进行一致性聚类分析,将其分成Cluster1和Cluster2组,分析不同的临床特征和总体生存率在两组聚类中是否存在差异。
1.4 铜死亡相关预后基因的筛选及风险评估
使用“survival”软件包进行单因素Cox回归分析筛选预后相关基因,使用LASSO回归分析以消除假阳性预后相关铜死亡基因,使用“glmnet”和“survival”软件包,计算基因之间的相关回归系数,根据回归系数加权建立肝癌预后风险评估公式:

公式中n指基因数量,Expi表示每个基因的表达量,Coei表示回归系数。根据不同样品中特征基因的表达,为每例患者分配风险评分,根据评分中位值将患者分为高风险组和低风险组,通过Kaplan-Meier曲线和计算ROC曲线下的面积(AUC)评估风险评分的预后意义和诊断效能。将性别、年龄、TNM分期、组织学分级和风险评分纳入单因素和多因素Cox回归中,分析多种临床特征干扰下风险评分的预后独立性,并通过“forestplot”软件包的森林图可视化每个变量的P值、风险比(HR)和95%CI。
1.5 GO功能富集分析和KEGG信号通路分析
为了获得与疾病发生发展所涉及的生物学功能和信号通路,使用仙桃学术平台(https://www.xiantao.love/)进行注释和可视化,对5个铜死亡相关基因进行基因本体(GO)分析和京都基因组百科全书(KEGG)通路分析。
1.6 统计学方法
采用R语言(4.1.3)软件进行统计分析。采用Wilcoxon秩和检验进行两组之间基因差异分析。Spearman相关检验分析基因表达间的相关性。Kaplan-Meier法绘制生存曲线。ROC曲线评估预后模型的敏感度和特异性。P < 0.05为差异有统计学意义。
2 结果
2.1 铜死亡相关基因差异表达分析
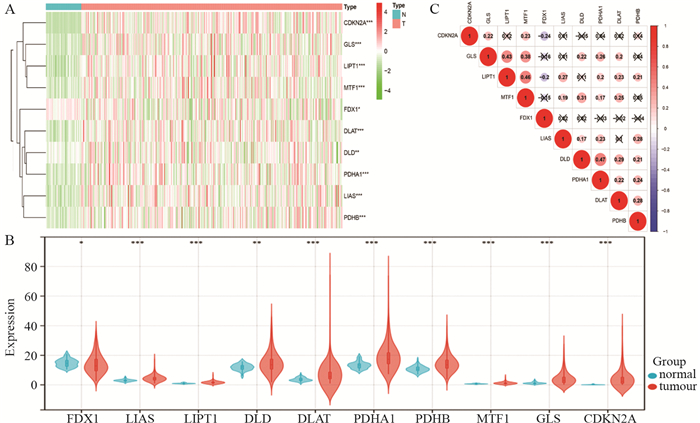
在TCGA队列中,肝癌组织和正常组织样本之间10个铜死亡相关基因的表达差异均有统计学意义(P < 0.05),其中FDX1在肝癌组织中下调,其余9个基因表达上调,见图 1A~B。相关性分析显示,大部分铜死亡相关基因都呈正相关,其中LIPT1与MTF1呈现最强正相关(r=0.46);而FDX1与CDKN2A(r=-0.24),FDX1与LIPT1(r=-0.2)呈显著负相关,见图 1C。
![]() 图 1 铜死亡相关基因在肝癌中的表达水平及其相关性Figure 1 Expression level and correlation of cuprotosis-related genes in liver cancerA: expression levels of 10 cuprotosis-related genes marked with an asterisk in the heat map were was not significant, *: P < 0.05, **: P < 0.01, ***: P < 0.001); B: violin plot of the expression levels of 10 cuprotosis-related genes in liver cancer and normal control samples; C: Spearman correlation analysis of 10 genes related to cuprotosis in liver cancer samples.
图 1 铜死亡相关基因在肝癌中的表达水平及其相关性Figure 1 Expression level and correlation of cuprotosis-related genes in liver cancerA: expression levels of 10 cuprotosis-related genes marked with an asterisk in the heat map were was not significant, *: P < 0.05, **: P < 0.01, ***: P < 0.001); B: violin plot of the expression levels of 10 cuprotosis-related genes in liver cancer and normal control samples; C: Spearman correlation analysis of 10 genes related to cuprotosis in liver cancer samples.2.2 一致性聚类分析铜死亡相关基因
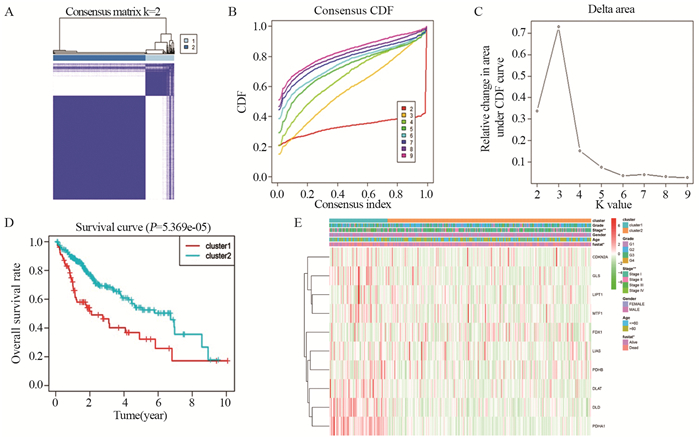
根据铜死亡相关基因的表达谱,对TCGA数据库中371例肝癌样本进行聚类分析。当k=2时,肝癌样本组内相关性最高,组间相关性较低,聚类效果稳定,确定了2个肝癌亚型,见图 2A~C。Cluster1的生存时间明显短于Cluster2(P < 0.05),见图 2D。此外,Cluster1与诊断时较高的分期(P < 0.01)和较高的死亡状态(P < 0.05)显著相关,而与肿瘤分级、年龄、性别无关(P > 0.05),见图 2E。
![]() 图 2 一致性聚类分析铜死亡相关基因Figure 2 Consistent cluster analysis of cuprotosis-related genesA: k=2 consistency clustering matrix; B: k=2–9 consistent clustering cumulative distribution function (CDF); C: The relative change in area under CDF curve when k=2–9; D: The overall survival rate in the clustering of the two groups; E: the relationship between different clustering and clinicopathology; P values were marked with an asterisk, **: P < 0.01.
图 2 一致性聚类分析铜死亡相关基因Figure 2 Consistent cluster analysis of cuprotosis-related genesA: k=2 consistency clustering matrix; B: k=2–9 consistent clustering cumulative distribution function (CDF); C: The relative change in area under CDF curve when k=2–9; D: The overall survival rate in the clustering of the two groups; E: the relationship between different clustering and clinicopathology; P values were marked with an asterisk, **: P < 0.01.2.3 风险预测模型的建立与评估
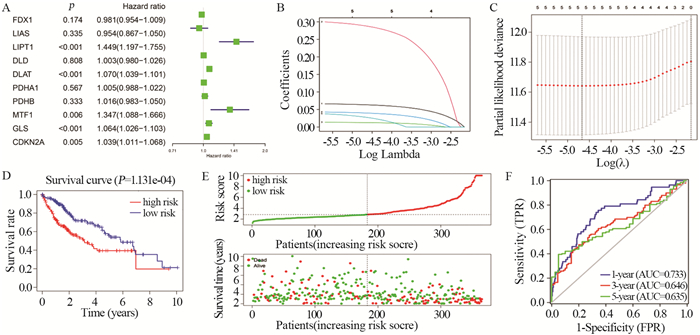
移除所有生存时间未知且无生存状态的样本后,共有365例肝癌样本与具有完整生存信息的相应患者匹配。将所有铜死亡相关基因纳入单因素Cox回归模型中,筛选得到5个基因(LIPT1、DLAT、MTF1、GLS、CDKN2A)均为HR > 1(且P < 0.05)的危险基因,见图 3A。通过LASSO回归分析进一步减少风险模型中包含的基因数量,最终5个基因全部被纳入风险模型中,见图 3B~C。风险评分计算如下:风险评分=(0.293×LIPT1exp)+(0.064×DLATexp)+(0.031×MTF1exp)+(0.013×GLSexp)+(0.041×CDKN2Aexp)。根据风险评分公式计算的中位值将患者分为高危组和低危组。Kaplan-Meier生存曲线结果表明高危组在肝癌患者中的生存率较差,见图 3D,随着风险评分的上升,死亡的患者例数明显上升(P < 0.05),这表明风险评分和生存时间显著相关,见图 3E。ROC曲线分析显示该模型预测肝癌患者术后1、3、5年生存率的AUC值分别为0.733、0.646、0.635,见图 3F。
![]() 图 3 筛选独立预后因素Figure 3 Screening of independent prognostic factorsA: ten cuprotosis prognostic genes were screened by univariate Cox regression, and HR and 95%CI were calculated; B: LASSO regression coefficient distribution diagram; C: the optimal λ value was selected through cross validation; D: survival status and survival distribution; E: survival curves of high-risk and low-risk groups; F: the predictive effectiveness of the risk model was evaluated with the ROC curve.
图 3 筛选独立预后因素Figure 3 Screening of independent prognostic factorsA: ten cuprotosis prognostic genes were screened by univariate Cox regression, and HR and 95%CI were calculated; B: LASSO regression coefficient distribution diagram; C: the optimal λ value was selected through cross validation; D: survival status and survival distribution; E: survival curves of high-risk and low-risk groups; F: the predictive effectiveness of the risk model was evaluated with the ROC curve.2.4 风险预测模型的独立预后价值
单因素Cox回归分析结果表明,风险评分(HR=1.146, P < 0.001)和分期(HR=1.680, P < 0.001)是肝癌恶化的危险因素,见图 4A。多因素Cox分析结果表明,在调整其他混杂因素后,风险评分(HR=1.117, P < 0.001)仍是一个独立的预后因素,见图 4B。计算风险评分的AUC值为0.727,明显高于其他临床特征的AUC值,见图 4C,表明5个铜死亡相关基因对肝癌的预后风险预测模型可靠。高危组和低危组患者中的5个铜死亡相关基因在肝癌的分级和分期之间存在统计学差异(P < 0.01),见图 4D。
![]() 图 4 风险预测模型的独立预后价值Figure 4 Independent prognostic value of risk prediction modelsA: Univariate Cox regression analysis of risk score and clinical characteristic parameters; B: Multivariate Cox regression analysis of risk score and clinical characteristic parameters; C: ROC curve analysis of risk score; D: Expression levels and clinical characteristic distribution of five copper death-related genes in low-risk and high-risk HCC patients, P values were marked with asterisk, **: P < 0.01.
图 4 风险预测模型的独立预后价值Figure 4 Independent prognostic value of risk prediction modelsA: Univariate Cox regression analysis of risk score and clinical characteristic parameters; B: Multivariate Cox regression analysis of risk score and clinical characteristic parameters; C: ROC curve analysis of risk score; D: Expression levels and clinical characteristic distribution of five copper death-related genes in low-risk and high-risk HCC patients, P values were marked with asterisk, **: P < 0.01.2.5 GO功能富集分析和KEGG信号通路分析
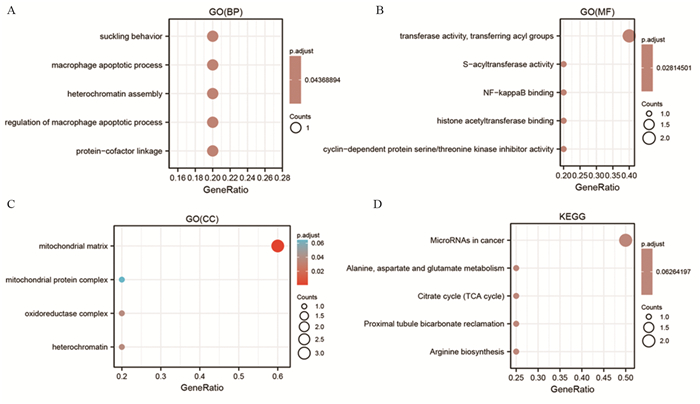
在生物学过程(BP)方面,5个铜死亡基因主要在巨噬细胞凋亡过程及其调控、异染色质组装等方面富集,见图 5A。对于分子功能(MF),铜死亡基因在转移酶活性、酰基转移酶活性等方面富集,见图 5B。对于细胞成分(CC),铜死亡基因在线粒体基质、线粒体蛋白复合物以及氧化还原酶复合物等方面富集,见图 5C。在KEGG途径富集分析中,铜死亡基因主要与丙氨酸、天冬氨酸和谷氨酸的代谢以及柠檬酸循环(TCA循环)有关,见图 5D。
![]() 图 5 铜死亡相关基因的GO和KEGG富集分析Figure 5 GO and KEGG functional enrichment analysis of cuprotosis-related genesA: biological processes(BP) in GO analysis; B: molecular function(MF) in GO analysis; C: cellular components(CC) in GO analysis; D: KEGG pathways analysis.
图 5 铜死亡相关基因的GO和KEGG富集分析Figure 5 GO and KEGG functional enrichment analysis of cuprotosis-related genesA: biological processes(BP) in GO analysis; B: molecular function(MF) in GO analysis; C: cellular components(CC) in GO analysis; D: KEGG pathways analysis.3 讨论
本研究拟探讨铜死亡相关基因在肝癌中的作用,发现其与肝癌的临床特征密切相关。首先,我们分析了10个目前已知的铜死亡相关基因的mRNA水平在肝癌组织和正常组织中的表达量,发现全部基因的表达量都存在差异。然后,基于这些差异表达的基因进行一致性聚类分析,确定了肝癌的两个亚组,第1亚组的预后较差。为了进一步评估这些铜死亡相关基因的预后价值,通过单因素Cox回归分析和LASSO回归分析构建了5个与铜死亡相关基因(LIPT1、DLAT、MTF1、GLS和CDKN2A)的风险特征模型,研究表明风险评分不仅是一个独立的预后指标,而且可以预测肝癌的临床特征。此外,功能分析显示,与TCA循环相关的通路被富集。因此,利用5个铜死亡相关基因建立的肝癌预后模型是有意义的。
DLAT、MTF1、GLS、CDKN2A属于丙酮酸脱氢酶复合体(PDC)中编码蛋白的基因,该复合体是丙酮酸进入线粒体后转化为乙酰-CoA的唯一途径。而二氢硫辛酰胺转乙酰基酶基因(DLAT)属于PDC E2亚单位,Goh等[9]发现DLAT在胃癌细胞中表达显著上调,Shan等[10]发现DLAT通过促进戊糖磷酸途径的激活进而促进肿瘤细胞生长。金属调控转录因子1基因(MTF1)是一种锌指转录因子,通过激活下游靶基因促进细胞存活。Ji等[11]研究表明,MTF1具有致癌作用,并通过促进上皮—间充质转化从而促进卵巢肿瘤转移。在p53存在的乳腺癌细胞中,MTF1可被锌和铜激活。MTF1可能是一种新的早期诊断生物标志物,也是临床治疗的药物靶点。谷氨酰胺酶基因(GLS)转化为三羧酸循环代谢产物是促进癌细胞增殖的关键代谢过程,已被确定为癌症代谢的标志[12]。Guo等[13]发现GLS的表达在子宫内膜癌进展过程中升高,并与不良预后相关。Zhang等[14]发现线粒体GCN5L1通过调节GLS乙酰化和活性在肝癌发展过程中发挥肿瘤调节器的作用。细胞周期依赖性激酶抑制基因(CDKN2A)又称为P16基因,是一种多肿瘤抑制基因,能够参与细胞周期的调控,抑制细胞增殖和分裂,而肿瘤细胞增殖速度较正常细胞快,这可能是因为肿瘤细胞CDKN2A基因突变或失活,从而使其增殖分化速度增加。而关于脂酰转移酶1基因(LIPT1)的相关报道较少,其突变可引起丙酮酸和α-酮戊二酸脱氢酶继发缺乏的Leigh病,这是一种是由于呼吸链亚单位缺失造成的先天性代谢紊乱性疾病,是一种线粒体脑肌病[15]。铜死亡观点的提出及进一步揭示的具体机制,将为铜失调疾病、部分大量表达脂酰化线粒体蛋白及具有高度呼吸作用的癌症等疾病提供新的治疗策略。
本研究有一定局限性:首先,使用的是公共数据库的回顾性数据,缺乏前瞻性临床数据来验证其预后意义;其次,考虑到肝癌是一种典型的多基因疾病,本文仅用5个铜死亡相关基因来建立预后模型,可能缺少其他能更精准预测肝癌预后的基因。
综上所述,本研究使用5个铜死亡相关基因构建了一个新的风险评分模型,并且与生存时间独立相关,为预测肝癌预后提供了新的见解。
Competing interests: The authors declare that they have no competing interests.作者贡献:赵小乐:数据分析与文稿撰写钱波:文章修改邵泉:提供研究思路 -
![]()
图 1 铜死亡相关基因在肝癌中的表达水平及其相关性
Figure 1 Expression level and correlation of cuprotosis-related genes in liver cancer
-
[1] Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries[J]. CA Cancer J Clin, 2021, 71(3): 209-249. doi: 10.3322/caac.21660
[2] Chen Z, Xie H, Hu M, et al. Recent progress in treatment of hepatocellular carcinoma[J]. Am J Cancer Res, 2020, 10(9): 2993-3036.
[3] Xiao S, Huang S, Yang J. Overexpression of GIHCG is Associated with a Poor Prognosis and Immune Infiltration in Hepatocellular Carcinoma[J]. Onco Targets Ther, 2020, 13: 11607-11619. doi: 10.2147/OTT.S271966
[4] Chen X, Wang L, Hong L, et al. Identification of Aging-Related Genes Associated With Clinical and Prognostic Features of Hepatocellular Carcinoma[J]. Front Genet, 2021, 12: 661988. doi: 10.3389/fgene.2021.661988
[5] Liang JY, Wang DS, Lin HC, et al. A Novel Ferroptosis-related Gene Signature for Overall Survival Prediction in Patients with Hepatocellular Carcinoma[J]. Int J Biol Sci, 2020, 16(13): 2430-2441. doi: 10.7150/ijbs.45050
[6] Blockhuys S, Celauro E, Hildesjö C, et al. Defining the human copper proteome and analysis of its expression variation in cancers[J]. Metallomics, 2017, 9(2): 112-123. doi: 10.1039/C6MT00202A
[7] Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins[J]. Science, 2022, 375(6586): 1254-1261. doi: 10.1126/science.abf0529
[8] 龚琳婧, 石毓君. 癌症中调节性细胞死亡方式的研究进展与未来展望[J]. 中国普外基础与临床杂志, 2022, 27(5): 582-584. https://www.cnki.com.cn/Article/CJFDTOTAL-ZPWL202205020.htm Gong LJ, Shi YJ. Progress and prospects: regulated cell death in cancer[J]. Zhongguo Pu Wai Ji Chu Yu Lin Chuang Za Zhi, 2022, 27(5): 582-584. https://www.cnki.com.cn/Article/CJFDTOTAL-ZPWL202205020.htm
[9] Goh WQ, Ow GS, Kuznetsov VA, et al. DLAT subunit of the pyruvate dehydrogenase complex is upregulated in gastric cancer-implications in cancer therapy[J]. Am J Transl Res, 2015, 7(6): 1140-1151.
[10] Shan C, Elf S, Ji Q, et al. Lysine acetylation activates 6-phosphogluconate dehydrogenase to promote tumor growth[J]. Mol Cell, 2014, 55(4): 552-565. doi: 10.1016/j.molcel.2014.06.020
[11] Ji L, Zhao G, Zhang P, et al. Knockout of MTF1 Inhibits the Epithelial to Mesenchymal Transition in Ovarian Cancer Cells[J]. J Cancer, 2018, 9(24): 4578-4585. doi: 10.7150/jca.28040
[12] Yang L, Venneti S, Nagrath D. Glutaminolysis: A Hallmark of Cancer Metabolism[J]. Annu Rev Biomed Eng, 2017, 19: 163-194. doi: 10.1146/annurev-bioeng-071516-044546
[13] Guo H, Li W, Pan G, et al. The Glutaminase Inhibitor Compound 968 Exhibits Potent in vitro and in vivo Anti-tumor Effects in Endometrial Cancer[J]. Anticancer Agents Med Chem, 2022. Online ahead of print.
[14] Zhang T, Cui Y, Wu Y, et al. Mitochondrial GCN5L1 regulates glutaminase acetylation and hepatocellular carcinoma[J]. Clin Transl Med, 2022, 12(5): e852.
[15] Soreze Y, Boutron A, Habarou F, et al. Mutations in human lipoyltransferase gene LIPT1 cause a Leigh disease with secondary deficiency for pyruvate and alpha-ketoglutarate dehydrogenase[J]. Orphanet J Rare Dis, 2013, 8: 192. doi: 10.1186/1750-1172-8-192
-
期刊类型引用(1)
1. 吴晓昀,荆林林,齐夏丽. 2型糖尿病合并乳腺癌患者预后高危因素分析. 实用癌症杂志. 2024(08): 1356-1358 .  百度学术
百度学术
其他类型引用(0)
 下载:
下载:

计量
- 文章访问数: 1651
- HTML全文浏览量: 695
- PDF下载量: 840
- 被引次数: 1

